基于cd-ag共掺杂改善zno单层光催化特性的仿真方法
技术领域
1.本发明属于半导体技术领域,特别涉及一种改善zno单层光催化特性的仿真方法,可用于zno单层材料的理论模拟和光催化应用。
背景技术:
2.随着工业革命的进一步发展,能源的枯竭以及环境污染严重影响着人们的生活,半导体材料光催化剂可以有效的降解污染物,从而引起了人们的广泛关注。zno作为一种ii-vi化合物半导体,具有化学稳定性高、载流子迁移率高、激子结合能大(60mev)、合成技术成熟、性能可调等特点,使其在众多的光催化半导体材料中脱颖而出。与zno体材料相比,二维纳米材料zno单层具有更独特的性能,如更大的表面积、更多的光催化反应位点、更短的光诱导载流子传输路径等,这些都有利于提高光催化性能,但在实际应用中存在太阳光利用率低、光生载流子重新复合降低催化活性和催化剂回收利用难等缺点,限制了其进一步的发展。
3.近年来,人们通过在zno材料中引入杂质、缺陷来改变其形貌等方式来拓展其光谱响应范围。
4.徐殿双等人在“单层zno及其掺杂的第一性原理”文章中开展了cd、ag的单掺杂zno单层的研究,研究发现掺杂cd的zno单层的禁带宽度减小,并且随着cd浓度的增加,介电函数进一步往低能方向转移,发生红移现象导致cd掺杂zno单层的可见光吸收系数增加,提高了光催化活性,此外ag掺杂zno单层,禁带宽度同样减小,介电函数红移,禁带中引入了杂质能级,但是这种用cd和ag的单掺杂对zno单层光催化性能的提高并不明显,从而使得在实际中对于污水的有机污染物的降解效率较低,处理不彻底,容易造成二次污染。
5.吴昌瀚等人在“cd-ag共掺杂zno及cd-ag:zno/cu2o光伏电池的模拟研究”文章中开展了cd-ag共掺杂zno体材料的理论研究,他们的工作集中于cd-ag共掺杂对于zno体材料能带结构的影响,没有开展光催化特性方面的研究。目前已有实验报道cd-ag共掺杂能够有效提高zno纳米材料的光催化性能,但是文章中对于zno材料的光催化性能的提升也不明显,且目前没有关cd-ag共掺杂zno单层光催化性能的文献报道。
技术实现要素:
6.本发明的目的在于针对上述现有技术存在的缺陷,提出一种基于cd-ag共掺杂改善zno单层光催化特性的仿真方法,以得到cd-ag共掺杂zno单层材料的光催化特性,使其应用到污水处理中,对污水中的有机污染物降解能力得到进一步提升,有效保护环境。
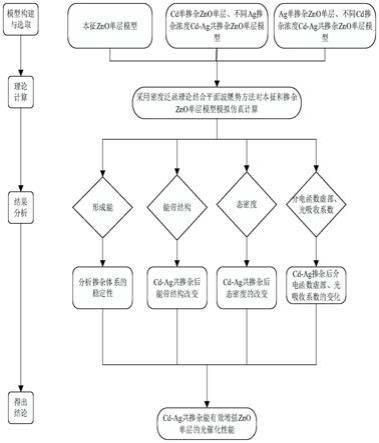
7.本发明的目的是通过下述技术方案实现的:
8.一、技术原理
9.zno是一种性能优异的半导体光催化剂,具有成本低,化学稳定性好,无毒,激子结合能高达60mev等优点,对有机污染物具有很高的光催化作用,但是其禁带宽度较大的特点限制了zno单层在光催化剂方面的应用。因此有必要开展提高zno单层材料光催化性能方面
的研究,为zno单层在半导体光催化领域的发展提供参考。
10.本发明通过构建本征zno单层、cd单掺杂zno单层、不同ag掺杂浓度的cd-ag共掺杂zno单层、ag单掺杂zno单层和不同cd掺杂浓度的cd-ag共掺杂zno单层模型,采用密度泛函理论结合平面波赝势方法计算本征zno单层、cd单掺杂zno单层、不同ag掺杂浓度的cd-ag共掺杂zno单层、ag单掺杂zno单层和不同cd掺杂浓度下cd-ag共掺杂zno单层模型的几何结构、形成能,能带结构、态密度和光学特性,通过对比分析,以实现cd-ag共掺杂对zno单层光催化性能的提高。
11.二.实现方案
12.根据上述原理,本发明提供一种基于cd-ag共掺杂改善zno单层光催化特性的仿真方法,其实现步骤:
13.1)将zno体材料原胞沿x轴和y轴构成的平面进行切片,在切片后的zno材料z轴方向上添加真空层得到zno单层,并对该zno单层进行几何优化,得到本征zno单层模型;
14.2)在本征zno单层模型中掺杂cd原子,得到cd单掺杂zno单层模型,并采用广义梯度近似法对该模型进行几何优化计算体系总能量e
total
,得到优化后的cd单掺杂zno单层;
15.3)在cd单掺杂zno单层模型的不同掺杂位置掺杂不同浓度的ag,并进行几何优化和总能量e
total
选取,得到不同ag掺杂浓度的cd-ag共掺杂zno单层模型;
16.4)在本征zno单层模型中掺杂ag原子,得到ag单掺杂zno单层模型,并采用广义梯度近似法对该模型进行几何优化计算体系总能量e
total
,得到优化后的ag单掺杂zno单层模型;
17.5)在ag单掺杂zno单层模型的不同掺杂位置掺杂不同浓度的cd,并进行几何优化和总能量e
total
选取,得到不同cd掺杂浓度的cd-ag共掺杂zno单层模型;
18.6)根据上述2)、3)、4)、5)各步优化后的总能量e
total
,计算掺杂体系的形成能e
form
;
19.7)采用密度泛函理论结合平面波赝势方法对1)、2)、3)、4)、5)各步几何优化后的模型进行仿真计算,得到能带结构,态密度和光学特性:
20.7a)对1)、2)、3)、4)、5)各步几何优化后的模型的各个原子的电子轨道增加一个能量项进行库伦修正,得到库伦修正后的模型,采用广义梯度近似法加上库伦修正的方法对库伦修正后的的模型进行能量优化,计算得到本征和掺杂zno单层的能带结构图,态密度图和光学特性;
21.7b)通过对6)中计算的形成能e
form
和7a)中得到的能带结构图,态密度图和光学特性的分析对比,得到通过cd-ag共掺杂提高zno单层的光催化特性。
22.本发明与现有技术相比具有如下有益效果:
23.1、本发明通过模型构建、参数的设置实现本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和cd-ag共掺杂zno单层的几何结构、能带结构图、态密度图和光学特性的理论模拟,相较于现有技术通过实验验证,既能够得到较为精准的结果,同时避免了实验过程中多种不确定因素导致的人力财力的浪费;
24.2、本发明通过对于本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和cd-ag共掺杂zno单层能带结构图,态密度图和光学特性的计算对比,得到cd-ag共掺杂能够有效地提高zno单层的光催化性能,从而实现对于污水中的有机污染物降解能力的提升。
附图说明
25.为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍。
26.图1是本发明的实现流程示意图;
27.图2是本发明中zno单层优化前后的侧视示意图与优化后的俯视示意图;
28.图3是本发明中cd单掺zno单层和不同ag掺杂浓度cd-ag共掺杂的zno单层模型示意图;
29.图4是本发明中ag单掺zno单层和不同cd掺杂浓度cd-ag共掺杂的zno单层模型示意图;
30.图5是本发明中对掺杂zno单层进行几何优化后的结构示意图;
31.图6是本发明中cd单掺zno单层,不同ag掺杂浓度cd-ag共掺杂zno单层,ag单掺zno单层和不同cd掺杂浓度cd-ag共掺杂zno单层的形成能示意图;
32.图7是本发明中本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同ag掺杂浓度cd-ag共掺杂zno单层能带结构示意图;
33.图8是本发明中本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同ag掺杂浓度cd-ag共掺杂zno单层态密度示意图;
34.图9是本发明中本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同ag掺杂浓度cd-ag共掺杂zno单层介电函数虚部和光吸收系数示意图;
35.图10是本发明中不同cd掺杂浓度cd-ag共掺杂zno单层能带结构示意图;
36.图11是本发明中不同cd掺杂浓度cd-ag共掺杂zno单层态密度示意图;
37.图12是本发明中本征zno单层,cd单掺杂zno单层、ag单掺杂zno单层和不同cd掺杂浓度下cd-ag共掺杂zno单层的介电函数虚部和光吸收系数示意图。
具体实施方式
38.为使本发明的目的、技术方案和优点更加清楚,下面结合附图和具体实施方式进一步详细说明。
39.参照图1,本发明的实现步骤如下:
40.步骤1、对zno体材料进行切片,并对其进行添加真空层和几何优化,得到zno单层模型。
41.所述zno体材料在常温下为纤锌矿结构,其空间群为p63mc,晶格沿c轴呈六角形紧密排列,zno的晶格参数为三个键角分别是α=90
°
,β=90
°
,γ=120
°
,其原胞由两个zn原子和两个o原子构成。
42.本步骤的具体实现如下:
43.1.1)构建4
×4×
1的zno体材料原胞,沿x轴和y轴构成的平面进行切片,在切片后的zno材料z轴方向上添加的真空层得到zno单层模型,以避免周期性重复的膜层之间的相互作用;
44.1.2)对zno单层模型进行几何优化,得到4
×4×
1的本征zno单层模型:
45.1.2.1)设置参数:
46.通过广义梯度近似方法和修正泛函来描述电子间的交换关联能,通过超软赝势来
描述价电子与原子核之间的相互作用;
47.设k点网格3
×3×
1,布里渊区k点的路径为γ-α-η-k-γ-μ-l-h,平面波能量精度为1
×
10-5
ev/atom,原子之间相互作用力应小于晶体的内应力小于0.05gpa,原子的最大位移为自洽收敛误差设置为1
×
10-6
ev/atom,平面波截断能设定为400ev;
48.1.2.2)对zno单层模型的晶格常数和原子位置同时进行优化:
49.1.2.2a)选择优化超胞,设置迭代次数为100;
50.1.2.2b)在上述参数条件下进行第一步迭代,计算zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面;
51.1.2.2c)对势能面求最低极小值,由于此时该值大于设置的收敛精度,则该步迭代结束,改变晶格常数或移动原子位置进行1.2.2d)迭代计算;
52.1.2.2d)重新计算zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,将该值与设置的能量的收敛精度进行比较:
53.若该值小于设置的收敛精度,则整个迭代过程自动中断,几何优化结束;
54.若该值大于设置的收敛精度,则该步迭代结束,执行1.2.2e);
55.1.2.2e)改变zno单层模型的晶格常数或移动原子位置进行下一步迭代计算,重复该步操作,直到系统在设置的迭代次数内,检测到势能面的最低极小值小于设定的能量收敛精度时视为收敛,整个迭代过程自动中断,几何优化结束,得到zno单层优化前后的侧视示意图与优化后的俯视示意图,如图2所示。其中:
56.图2(a)是zno单层优化前的侧视图;
57.图2(b)和图2(c)是zno单层优化后的侧视图与俯视图;
58.从图2(a)中可以看出优化前zno单层平面褶皱不平,从图2(b),图2(c)中可以看出优化后所有原子位于同一个平面,呈平面六元环结构。
59.步骤2、在本征zno单层超胞模型中掺杂cd原子,得到cd单掺杂zno单层模型,并采用广义梯度近似法对该模型进行几何优化计算体系总能量e
total
,得到优化后的cd单掺杂zno单层。
60.本步骤的具体实现是:
61.2.1)在步骤1)构建的本征zno单层模型基础上,用一个cd原子替代zn原子得到cd单掺杂zno单层模型,如图3(a);
62.2.2)采用广义梯度近似法对cd单掺杂zno单层模型进行几何优化,得到优化后的cd单掺杂zno单层,记为cd-zno:
63.2.2.1)设置参数,其与步骤1.2.1)相同;
64.2.2.2)对cd单掺杂zno单层模型的晶格常数和原子位置同时进行优化:
65.2.2.2a)选择优化超胞,设置迭代次数为100;
66.2.2.2b)在上述参数条件下进行第一步迭代,计算cd单掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面;
67.2.2.2c)对势能面求最低极小值,由于此时该值大于设置的收敛精度,则该步迭代结束,改变晶格常数或移动原子位置进行2.2.2d)迭代计算;
68.2.2.2d)重新计算cd单掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,将该值与设置的能量的收敛精度进行比较:
69.若该值小于设置的收敛精度,则整个迭代过程自动中断,几何优化结束;
70.若该值大于设置的收敛精度,则该步迭代结束,执行2.2.2e);
71.2.2.2e)改变cd单掺杂zno单层模型的晶格常数或移动原子位置进行下一步迭代计算,重复该步操作,直到系统在设置的迭代次数内,检测到势能面的最低极小值小于设定的能量收敛精度时视为收敛,整个迭代过程自动中断,几何优化结束。
72.优化后的cd单掺杂zno单层体系总能量为-33922.6614ev,优化后的cd单掺杂zno单层的几何结构如图5(a)所示。
73.步骤3、在cd单掺杂zno单层模型的不同掺杂位置掺杂不同浓度的ag,并进几何优化和总能量e
total
选取,得到三种不同ag掺杂浓度的cd-ag共掺杂zno单层模型。
74.3.1)在cd单掺杂zno单层模型上,选择在距cd原子为近邻1、次近邻2、远近邻3三种不同掺杂位置上掺入ag原子,得到三种不同的cd:ag=1:1的cd-ag共掺杂zno单层模型,如图3(b)所示;
75.3.2)采用广义梯度近似法对三种不同的cd:ag=1:1的cd-ag共掺杂zno单层模型分别进行几何优化计算掺杂体系的总能量,选取总能量最低的掺杂模型:
76.3.2.1)对于ag掺杂在近邻位置1的cd:ag=1:1的cd-ag共掺杂zno单层模型进行几何优化,其步骤如下:
77.3.2.1a)设置参数,其与步骤1.2.1)相同;
78.3.2.1b)对ag掺杂在近邻位置1的cd:ag=1:1的cd-ag共掺杂zno单层模型的晶格常数和原子位置同时进行优化:
79.3.2.1b1)选择优化超胞,设置迭代次数为100;
80.3.2.1b2)在上述参数条件下进行第一步迭代,计算ag掺杂在近邻位置1的cd:ag=1:1的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面;
81.3.2.1b3)对势能面求最低极小值,由于此时该值大于设置的收敛精度,则该步迭代结束,改变晶格常数或移动原子位置进行3.2.1b4)迭代计算;
82.3.2.1b4)重新计算ag掺杂在近邻位置1的cd:ag=1:1的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,将该值与设置的能量的收敛精度进行比较:
83.若该值小于设置的收敛精度,则整个迭代过程自动中断,几何优化结束;
84.若该值大于设置的收敛精度,则该步迭代结束,执行3.2.1b5);
85.3.2.1b5)改变ag掺杂在近邻位置1的cd:ag=1:1的cd-ag共掺杂zno单层模型的晶格常数或移动原子位置进行下一步迭代计算,重复该步操作,直到系统在设置的迭代次数内,检测到势能面的最低极小值小于设定的能量收敛精度时视为收敛,整个迭代过程自动中断,几何优化结束。
86.3.2.2)对于ag掺杂在次近邻位置2的cd:ag=1:1的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤3.2.1)相同,只是该过程中优化的模型是ag掺杂在次近邻位
置2的cd:ag=1:1的cd-ag共掺杂zno单层模型。
87.3.2.3)对于ag掺杂在远近邻位置3的cd:ag=1:1的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤3.2.1)相同,只是该过程中优化的模型是ag掺杂在远近邻位置3的cd:ag=1:1的cd-ag共掺杂zno单层模型。
88.三种不同的cd:ag=1:1的cd-ag共掺杂zno单层模型优化后,其掺杂体系的总能量如表1所示:
89.表1三种不同的cd:ag=1:1的cd-ag共掺杂zno单层模型的总能量
90.ag掺杂位置123总能量e
total-33237.1396ev-33237.1013ev-33237.0501ev
91.从表1中可以得到,ag原子占据1位置时的总能量最低,因而选取在该位置用ag原子替代zn原子作为cd-ag-zno模型,该模型的几何结构如图5(b)所示;
92.3.3)在选取总能量最低的cd:ag=1:1的cd-ag共掺杂zno单层模型上,选择在距该模型确定的ag原子为近邻ⅰ、次近邻ⅱ和远近邻ⅲ三种不同掺杂位置上掺入ag原子,得到三种不同的cd:ag=1:2的cd-ag共掺杂zno单层模型,如图3(c)所示;
93.3.4)采用广义梯度近似法对三种不同的cd:ag=1:2的cd-ag共掺杂zno单层模型分别进行几何优化计算掺杂体系的总能量,选取总能量最低的掺杂模型:
94.3.4.1)对于ag掺杂在近邻位置ⅰ的cd:ag=1:2的cd-ag共掺杂zno单层模型进行几何优化,其步骤如下:
95.3.4.1a)设置参数,其与步骤1.2.1)相同;
96.3.4.1b)对ag掺杂在近邻位置ⅰ的cd:ag=1:2的cd-ag共掺杂zno单层模型的晶格常数和原子位置同时进行优化:
97.3.4.1b1)选择优化超胞,设置迭代次数为100;
98.3.4.1b2)在上述参数条件下进行第一步迭代,计算ag掺杂在近邻位置ⅰ的cd:ag=1:2的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面;
99.3.4.1b3)对势能面求最低极小值,由于此时该值大于设置的收敛精度,则该步迭代结束,改变晶格常数或移动原子位置进行3.4.1b4)迭代计算;
100.3.4.1b4)重新计算近邻位置ⅰ的cd:ag=1:2的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,将该值与设置的能量的收敛精度进行比较:
101.若该值小于设置的收敛精度,则整个迭代过程自动中断,几何优化结束;
102.若该值大于设置的收敛精度,则该步迭代结束,执行3.4.1b5);
103.3.4.1b5)改变ag掺杂在近邻位置ⅰ的cd:ag=1:2的cd-ag共掺杂zno单层模型的晶格常数或移动原子位置进行下一步迭代计算,重复该步操作,直到系统在设置的迭代次数内,检测到势能面的最低极小值小于设定的能量收敛精度时视为收敛,整个迭代过程自动中断,几何优化结束;
104.3.4.2)对于ag掺杂在次近邻位置ⅱ的cd:ag=1:2的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤3.4.1)相同,只是该过程中优化的模型是ag掺杂在次近邻位置ⅱ的cd:ag=1:2的cd-ag共掺杂zno单层模型;
105.3.4.3)对于ag掺杂在远近邻位置ⅲ的cd:ag=1:2的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤3.4.1)相同,只是该过程中优化的模型是ag掺杂在远近邻位置ⅲ的cd:ag=1:2的cd-ag共掺杂zno单层模型;
106.三种不同的cd:ag=1:2的cd-ag共掺杂zno单层模型优化后,其掺杂体系的总能量如表2所示:
107.表2三种不同的cd:ag=1:2的cd-ag共掺杂zno单层模型的总能量
108.ag掺杂位置
ⅰⅱⅲ
总能量e
total-32551.9206ev-32551.6900ev-32551.8328ev
109.从表2中可以得到,ag原子占据ⅰ位置时总能量最低,因而选取在该位置用ag原子替代zn原子作为cd-2ag-zno模型,该模型的几何结构如图5(c)所示;
110.3.5)在选取总能量最低的cd:ag=1:2的cd-ag共掺杂zno单层模型上,选择在距该模型确定的ag原子为近邻a、次近邻b和远近邻c三种不同掺杂位置上掺入ag原子,得到三种不同的cd:ag=1:3的cd-ag的cd-ag共掺杂zno单层模型,如图3(d)所示;
111.3.6)采用广义梯度近似法对三种不同的cd:ag=1:3的cd-ag共掺杂zno单层模型分别进行几何优化计算掺杂体系的总能量,选取总能量最低的掺杂模型:
112.3.6.1)对ag掺杂在近邻位置a的cd:ag=1:3的cd-ag共掺杂zno单层模型进行几何优化,其步骤如下:
113.3.6.1a)设置参数,其与步骤1.2.1)相同;
114.3.6.1b)对ag掺杂在近邻位置a的cd:ag=1:3的cd-ag共掺杂zno单层模型的晶格常数和原子位置同时进行优化:
115.3.6.1b1)选择优化超胞,设置迭代次数为100;
116.3.6.1b2)在上述参数条件下进行第一步迭代,计算ag掺杂在近邻位置a的cd:ag=1:3的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面;
117.3.6.1b3)对势能面求最低极小值,由于此时该值大于设置的收敛精度,则该步迭代结束,改变晶格常数或移动原子位置进行3.6.1b4)迭代计算;
118.3.6.1b4)重新计算ag掺杂在近邻位置a的cd:ag=1:3共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,将该值与设置的能量的收敛精度进行比较:
119.若该值小于设置的收敛精度,则整个迭代过程自动中断,几何优化结束;
120.若该值大于设置的收敛精度,则该步迭代结束,执行3.6.1b5);
121.3.6.1b5)改变ag掺杂在近邻位置a的cd:ag=1:3的cd-ag共掺杂zno单层模型的晶格常数或移动原子位置进行下一步迭代计算,重复该步操作,直到系统在设置的迭代次数内,检测到势能面的最低极小值小于设定的能量收敛精度时视为收敛,整个迭代过程自动中断,几何优化结束;
122.3.6.2)对于ag掺杂在次近邻位置b的cd:ag=1:3的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤3.6.1)相同,只是该过程中优化的模型是ag掺杂在次近邻位置b的cd:ag=1:3的cd-ag共掺杂zno单层模型;
123.3.6.3)对于ag掺杂在远近邻位置c的cd:ag=1:3的cd-ag共掺杂zno单层模型进行
几何优化,其优化过程与步骤3.6.1)相同,只是该过程中优化的模型是ag掺杂在远近邻位置c的cd:ag=1:3的cd-ag共掺杂zno单层模型。
124.三种不同的cd:ag=1:3的cd-ag共掺杂zno单层模型优化后,其掺杂体系的总能量如表3所示:
125.表3三种不同的cd:ag=1:3的cd-ag共掺杂zno单层模型的总能量
126.ag掺杂位置abc总能量e
total-31866.6364ev-31867.1116ev-31866.4680ev
127.从表3中可以得到,ag原子占据b位置时总能量最低,因而选取在该位置用ag原子替代zn原子作为cd-3ag-zno模型cd:ag=1:1,该模型的几何结构如图5(d)所示。
128.步骤4、在本征zno单层模型中掺杂ag原子,得到ag单掺杂zno单层模型,并采用广义梯度法对该模型进行几何优化计算体系总能量e
total
,得到优化后的ag单掺杂zno单层。
129.本步骤的具体实现是:
130.4.1)在步骤1)构建的本征zno单层模型基础上,用一个ag原子替代zn原子得到ag单掺杂zno单层模型,如图4(a);
131.4.2)并采用广义梯度法对该模型进行几何优化计算体系总能量e
total
,得到优化后的ag单掺杂zno单层,记为ag-zno:
132.4.2.1)设置参数,其与步骤1.2.1)相同;
133.4.2.2)对ag单掺杂zno单层模型的晶格常数和原子位置同时进行优化:
134.4.2.2a)选择优化超胞,设置迭代次数为100;
135.4.2.2b)在上述参数条件下进行第一步迭代,计算ag单掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面;
136.4.2.2c)对势能面求最低极小值,由于此时该值大于设置的收敛精度,则该步迭代结束,改变晶格常数或移动原子位置进行4.2.2d)迭代计算;
137.4.2.2d)重新计算ag单掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,将该值与设置的能量的收敛精度进行比较:
138.若该值小于设置的收敛精度,则整个迭代过程自动中断,几何优化结束;
139.若该值大于设置的收敛精度,则该步迭代结束,执行4.2.2e);
140.4.2.2e)改变ag单掺杂zno单层模型的晶格常数或移动原子位置进行下一步迭代计算,重复该步操作,直到系统在设置的迭代次数内,检测到势能面的最低极小值小于设定的能量收敛精度时视为收敛,整个迭代过程自动中断,几何优化结束。
141.优化后的ag单掺杂zno单层的体系总能量为-33664.1198ev,优化后的ag单掺杂zno单层的几何结构如图5(e)所示。
142.步骤5、在ag单掺杂zno单层模型的不同掺杂位置掺杂不同浓度的cd原子,并进行几何优化和总能量e
total
选取,得到不同cd掺杂浓度的cd-ag共掺杂zno单层模型。
143.5.1)在ag单掺杂zno单层模型上,选择在距ag原子为近邻1’、次近邻2’、远近邻3’三种不同掺杂位置上掺入cd原子,得到三种不同cd:ag=1:1的cd-ag共掺杂zno单层模型,如图4(b)所示;
144.5.2)采用广义梯度近似法对三种不同cd:ag=1:1的cd-ag共掺杂zno单层模型分
别进行几何优化计算掺杂体系的总能量,选取总能量最低的掺杂模型:
145.5.2.1)对于cd掺杂在近邻位置1’的cd:ag=1:1的cd-ag共掺杂zno单层模型进行几何优化,其步骤如下:
146.5.2.1a)设置参数,其与步骤1.2.1)相同;
147.5.2.1b)对cd掺杂在近邻位置1’的cd:ag=1:1的cd-ag共掺杂zno单层模型的晶格常数和原子位置同时进行优化:
148.5.2.1b1)选择优化超胞,设置迭代次数为100;
149.5.2.1b2)在上述参数条件下进行第一步迭代,计算cd掺杂在近邻位置1’的cd:ag=1:1的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面;
150.5.2.1b3)对势能面求最低极小值,由于此时该值大于设置的收敛精度,则该步迭代结束,改变晶格常数或移动原子位置进行5.2.1b4)迭代计算;
151.5.2.1b4)重新计算cd掺杂在近邻位置1’的cd:ag=1:1的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,将该值与设置的能量的收敛精度进行比较:
152.若该值小于设置的收敛精度,则整个迭代过程自动中断,几何优化结束;
153.若该值大于设置的收敛精度,则该步迭代结束,执行5.2.1b5);
154.5.2.1b5)改变cd掺杂在近邻位置1’的cd:ag=1:1的cd-ag共掺杂zno单层模型的晶格常数或移动原子位置进行下一步迭代计算,重复该步操作,直到系统在设置的迭代次数内,检测到势能面的最低极小值小于设定的能量收敛精度时视为收敛,整个迭代过程自动中断,几何优化结束。
155.5.2.2)对于cd掺杂在次近邻位置2’的cd:ag=1:1的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤5.2.1)相同,只是该过程中优化的模型是cd掺杂在次紧邻位置2’的cd:ag=1:1的cd-ag共掺杂zno单层模型。
156.5.2.3)对于cd掺杂在远近邻位置3’的cd:ag=1:1的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤5.2.1)相同,只是该过程中优化的模型是cd掺杂在远近邻位置3’的cd:ag=1:1的cd-ag共掺杂zno单层模型。
157.三种不同的cd:ag=1:1的cd-ag共掺杂zno单层模型优化后,其掺杂体系的总能量如表4所示:
158.表4三种不同的cd:ag=1:1的cd-ag共掺杂zno单层模型的总能量
159.cd掺杂位置1’2’3’总能量e
total
—
33237.1394ev-33237.0883ev-33237.0892ev
160.从表4中可以得到,cd原子占据1’位置时总能量最低,因而选取在该位置用cd原子替代zn原子作为cd-ag-zno’模型,该模型的几何结构如图5(f)所示;
161.5.3)在选取总能量最低的cd:ag=1:1的cd-ag共掺杂zno单层模型上,选择在距该模型确定的cd原子为近邻ⅰ’、次近邻ⅱ’和远近邻ⅲ’三种不同掺杂位置上掺入cd原子,得到三种不同的cd:ag=2:1的cd-ag共掺杂zno单层模型,如图4(c)所示;
162.5.4)采用广义梯度近似法对三种不同的cd:ag=2:1的cd-ag共掺杂zno单层模型分别进行几何优化计算掺杂体系的总能量,选取总能量最低的掺杂模型:
163.5.4.1)对cd掺杂在近邻位置ⅰ’的cd:ag=2:1的cd-ag共掺杂zno单层模型进行几何优化,其步骤如下:
164.5.4.1a)设置参数:其与步骤1.2.1)相同;
165.5.4.1b)对cd掺杂在近邻位置ⅰ’的cd:ag=2:1的cd-ag共掺杂zno单层模型的晶格常数和原子位置同时进行优化:
166.5.4.1b1)选择优化超胞,设置迭代次数为100;
167.5.4.1b2)在上述参数条件下进行第一步迭代,计算cd掺杂在近邻位置ⅰ’的cd:ag=2:1的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面;
168.5.4.1b3)对势能面求最低极小值,由于此时该值大于设置的收敛精度,则该步迭代结束,改变晶格常数或移动原子位置进行5.4.1b4)迭代计算;
169.5.4.1b4)重新计算cd掺杂在近邻位置ⅰ’的cd:ag=2:1的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,将该值与设置的能量的收敛精度进行比较:
170.若该值小于设置的收敛精度,则整个迭代过程自动中断,几何优化结束;
171.若该值大于设置的收敛精度,则该步迭代结束,执行5.4.1b5);
172.5.4.1b5)改变cd掺杂在近邻位置ⅰ’的cd:ag=2:1的cd-ag共掺杂zno单层模型的晶格常数或移动原子位置进行下一步迭代计算,重复该步操作,直到系统在设置的迭代次数内,检测到势能面的最低极小值小于设定的能量收敛精度时视为收敛,整个迭代过程自动中断,几何优化结束;
173.5.4.2)对于cd掺杂在次近邻位置ⅱ’的cd:ag=2:1的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤5.4.1)相同,只是该过程中优化的模型是cd掺杂在次近邻位置ⅱ’的cd:ag=2:1的cd-ag共掺杂zno单层模型;
174.5.4.3)对于cd掺杂在远近邻位置ⅲ’的cd:ag=2:1的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤5.4.1)相同,只是该过程中优化的模型是cd掺杂在远近邻位置ⅲ’的cd:ag=2:1的cd-ag共掺杂zno单层模型;
175.三种不同的cd:ag=2:1的cd-ag共掺杂zno单层模型优化后,其掺杂体系的总能量如表5所示:
176.表5三种不同的cd:ag=2:1的cd-ag共掺杂zno单层模型的总能量
177.cd掺杂位置ⅰ’ⅱ’ⅲ’总能量e
total-32810.0191ev-32810.1085ev-32809.9707ev
178.从表5中可以得到,cd原子占据ⅱ’位置时总能量最低,因而选取在该位置用cd原子替代zn原子作为2cd-ag-zno模型,该模型的几何结构如图5(g)所示;
179.5.5)在选取总能量最低的cd:ag=2:1的cd-ag共掺杂zno单层模型上,选择在距该模型确定的cd原子为近邻a’、次近邻b’和远近邻c’三种不同掺杂位置上掺入cd原子,得到三种不同的cd:ag=3:1的cd-ag共掺杂zno单层模型,如图4(d)所示;
180.5.6)采用广义梯度近似法对三种不同的cd:ag=3:1的cd-ag共掺杂zno单层模型分别进行几何优化计算掺杂体系的总能量,选取总能量最低的掺杂模型:
181.5.6.1)对cd掺杂在近邻位置a’的cd:ag=3:1的cd-ag共掺杂zno单层模型进行几
何优化,其步骤如下:
182.5.6.1a)设置参数:其与步骤1.2.1)相同;
183.5.6.1b)对cd掺杂在近邻位置a’的cd:ag=3:1的cd-ag共掺杂zno单层模型的晶格常数和原子位置同时进行优化:
184.5.6.1b1)选择优化超胞,设置迭代次数为100;
185.5.6.1b2)在上述参数条件下进行第一步迭代,计算cd掺杂在近邻位置a’的cd:ag=3:1的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面;
186.5.6.1b3)对势能面求最低极小值,由于此时该值大于设置的收敛精度,则该步迭代结束,改变晶格常数或移动原子位置进行5.6.1b4)迭代计算;
187.5.6.1b4)重新计算cd掺杂在近邻位置a’的cd:ag=3:1的cd-ag共掺杂zno单层模型的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,将该值与设置的能量的收敛精度进行比较:
188.若该值小于设置的收敛精度,则整个迭代过程自动中断,几何优化结束;
189.若该值大于设置的收敛精度,则该步迭代结束,执行5.6.1b5);
190.5.6.1b5)改变cd掺杂在近邻位置a’的cd:ag=3:1的cd-ag共掺杂zno单层模型的晶格常数或移动原子位置进行下一步迭代计算,重复该步操作,直到系统在设置的迭代次数内,检测到势能面的最低极小值小于设定的能量收敛精度时视为收敛,整个迭代过程自动中断,几何优化结束;
191.5.6.2)对于cd掺杂在次近邻位置b’的cd:ag=3:1的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤5.6.1)相同,只是该过程中优化的模型是cd掺杂在次近邻位置b’的cd:ag=3:1的cd-ag共掺杂zno单层模型;
192.5.6.3)对于cd掺杂在远近邻位置c’的cd:ag=3:1的cd-ag共掺杂zno单层模型进行几何优化,其优化过程与步骤5.6.1)相同,只是该过程中优化的模型是cd掺杂在远近邻位置c’的cd:ag=3:1的cd-ag共掺杂zno单层模型;
193.三种不同的cd:ag=3:1的cd-ag共掺杂zno单层模型优化后,其掺杂体系的总能量如表6所示:
194.表6三种不同的cd:ag=3:1的cd-ag共掺杂zno单层模型的总能量
195.cd掺杂位置a’b’c’总能量e
total-32383.0402ev-32383.2331ev-32382.9913ev
196.从表6中可以得到,cd原子占据b’位置时总能量最低,因而选取在位置cd原子替代zn原子作为3cd-ag-zno模型,该模型的几何结构如图5(h)所示。
197.步骤6、根据上述步骤2-5各步骤优化后的总能量计算掺杂体系的形成能e
form
。
198.e
form
=e
total-e
zno
+∑n
i
μ
i
199.其中e
total
是掺杂体系总能量,e
zno
表示的是本征zno单层的总能量,n
i
表示的是掺杂体系中得到或失去的原子个数,当n
i
小于零表示掺杂体系掺入原子,n
i
大于零表示的是掺杂体系失去原子,i表示的是掺入或失去的原子,μ
i
表示的是对应原子的化学势。在本实例中计算的掺杂体系的形成能如图6所示。从图6中可以看出从图中可以看出掺杂体系的形成能为正值,表明掺杂体系不易形成,随着ag/cd掺杂浓度的增大,共掺杂体系的形成能呈现
增长趋势,表明掺杂体系稳定性下降。
200.步骤7、采用密度泛函理论结合平面波赝势方法对步骤1、步骤2、步骤3、步骤4、步骤5各步骤几何优化后的模型进行仿真计算,得到能带结构,态密度和光学特性。
201.7.1)对步骤1、步骤2、步骤3,步骤4各步骤几何优化后的模型的各个原子的电子轨道增加一个能量项进行库伦修正,即zn的3d轨道、o的2p轨道、cd的4d轨道、ag的的4d轨道增加的能量项分别是10ev,7ev、2ev、5ev,得到库伦修正后的模型;
202.7.2)采用广义梯度近似加上库伦修正的方法对库伦修正后的模型进行能量优化,计算得到的本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同ag掺杂浓度cd-ag共掺杂zno单层的能带结构如图7所示,其中:
203.图7(a)是本征zno单层的能带结构,该图中给出了自旋向上和自旋向下的能带结构,该图中e
f
表示费米能级,从图中禁带宽度为4.062ev,其远大于纤锌矿结构的zno体材料的禁带宽度3.37ev,这主要是由于量子尺寸效应,两个自旋能带结构完全一致,表明本征zno单层不具备磁性;
204.图7(b)给出的是cd-zno的能带结构,图中给出了自旋向上和自旋向下的能带结构,该图中e
f
表示费米能级,从图中可以看出禁带宽度为3.866ev,其与本征zno单层相比禁带宽度减小。掺杂后导带底(cbm)和价带顶vbm位于γ点,两个自旋能带结构对称,表明cd-zno是无磁性直接带隙半导体材料,在-6ev附近出现了新的能级;
205.图7(c)是ag-zno的能带结构,该图中给出了自旋向上和自旋向下的能带结构,该图中e
f
表示费米能级,从图中可以看出两个自旋能带结构的不对称,表明ag-zno是具有磁性的,费米能级上方出现受主杂质能级。
206.图7(d)是cd-ag-zno的能带结构,该图中给出了自旋向上和自旋向下的能带结构,该图中e
f
表示费米能级,从图中可以看出费米能级靠近价带,表明掺杂体系属于p型掺杂,向上自旋与向下自旋能带结构的不对称,同时表明cd-ag共掺杂之后体系出现磁性。与cd-zno相比,随着ag元素的掺入在禁带中引入一条受主杂质能级,价带部分的能级分布较为密集,价带和导带都下移,且导带的下移程度更明显,因而禁带宽度变窄;
207.图7(e)是cd-2ag-zno的能带结构,该图中给出了自旋向上和自旋向下的能带结构,该图中e
f
表示费米能级,从图中可以看出相较于cd-ag-zno禁带宽度减小,向上自旋与向下自旋的能带结构对称,表明体系是无磁性的,同时表明cbm与vbm并不是位于同一高对称点上,因此cd-2ag-zno是间接带隙半导体材料,自旋向上的能带结构图中费米能级附近出现新的杂质能级;
208.图7(f)是cd-3ag-zno的能带结构,该图中给出了自旋向上和自旋向下的能带结构,该图中e
f
表示费米能级,从图中可以看出与cd-2ag-zno相比禁带宽度增加,向上自旋与向下自旋的能带结构是不对称的,表明该体系是具有磁性的,此外,一条新的杂质能级在向上自旋的能带结构图中费米能级附近被引入;
209.从图7(d)-7(f)的分析可以得到,对于cd-ag共掺杂zno单层体系,随着ag元素掺杂浓度的增加,cd-ag共掺杂zno单层的禁带宽度先减小后增大,禁带中引入更多的杂质能级,有利于电子的跃迁;
210.7.3)对步骤1、步骤2、步骤3,步骤4各步骤几何优化后的模型的各个原子的电子轨道增加一个能量项进行库伦修正,即zn的3d轨道、o的2p轨道、cd的4d轨道、ag的的4d轨道增
加的能量项分别是10ev,7ev、2ev、5ev,得到库伦修正后的模型;
211.7.4)采用广义梯度近似加上库伦修正的方法对步骤1、步骤2、步骤3,步骤4各步骤几何优化后的模型进行能量优化,计算得到的本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同ag掺杂浓度cd-ag共掺杂zno单层的态密度图如图8所示,其中:
212.图8(a)是本征zno单层的态密度图,该图中给出了自旋向上和自旋向下的总态密度tdos,自旋向上和自旋向下的分态密度pdos,从图中可以看出其价带主要是o-2p和zn-3d态的贡献,而导带主要由zn-3s态决定,此外在-14ev出现尖峰,主要来源于o-2s态;
213.图8(b)是cd-zno的态密度图,该图中给出了自旋向上和自旋向下的总态密度tdos,自旋向上和自旋向下的分态密度pdos,从图中可以看出其与本征zno单层态密度相比,导带向低能方向移动,禁带宽度减小,这与能带结构部分的分析保持一致,此外在-6ev附近出现一个态密度峰值,其主要来自于cd-4d轨道态;
214.图8(c)是ag-zno的态密度图,该图中给出了自旋向上和自旋向下的总态密度tdos,自旋向上和自旋向下的分态密度pdos,从图中可以看出tdos表现出上下不对称,表明ag掺杂体系是有磁性的。磁性的起源是ag-4d和o-2p。
215.图8(d)是cd-ag-zno的态密度图,该图中给出了自旋向上和自旋向下的总态密度tdos,自旋向上和自旋向下的分态密度pdos,从该图可以看出随着ag元素的掺入,总态密度总体向低能方向移动,总态密度表现出上下不对称,表明cd-ag共掺杂体系具有磁性的。导带主要是由ag-5s,o-2p和zn-4s三种轨道电子共同作用。价带顶主要由ag-4d和o-2p共同作用。o-2p轨道在1ev出现新的峰值,ag-4d在该位置处存在峰值大小相当的态密度峰,说明ag-4d和o-2p有较强的轨道杂化,这使得自旋向下的能带结构中出现一条杂质能级。在-14.6ev深能级处的态密度峰主要是o-2s和zn-3d的贡献;
216.图8(e)给出了cd-2ag-zno的态密度图,该图中给出了自旋向上和自旋向下的总态密度tdos,自旋向上和自旋向下的分态密度pdos,从图中可以看出其与cd-ag-zno相比,随着ag浓度的增加,态密度整体略微向高能方向移动,向上自旋的态密度图中1.41ev处出现了峰值,主要来自于ag-4d和o-2p的轨道杂化,这个峰值与自旋向上能带结构图中费米能级附近新增一条杂质能级相对应;
217.图8(f)给出了cd-3ag-zno-的态密度图,该图中给出了自旋向上和自旋向下的总态密度tdos,自旋向上和自旋向下的分态密度pdos,从图中可以看出ag元素浓度的进一步增大使得自旋向上态密度的费米能级附近o-2p峰值展宽增大与ag-4d态进行轨道杂化,从而使得费米能级附近的杂质能级展宽,这与能带结构部分分析的杂质能级增多的结论一致;
218.从图8(d)-8(f)分析可以得到,相比于本征zno单层和cd-zno的态密度,ag浓度的增大使得o-2p轨道出现新的态密度峰值,这些峰值与ag-4d发生轨道杂化,而这也是禁带中出现在杂质能级的来源;
219.7.5)对步骤1、步骤2、步骤3,步骤4各步骤几何优化后的模型的各个原子的电子轨道增加一个能量项进行库伦修正,即zn的3d轨道、o的2p轨道、cd的4d轨道、ag的的4d轨道增加的能量项分别是10ev,7ev、2ev、5ev,得到库伦修正后的模型;
220.7.6)采用广义梯度近似加上库伦修正的方法对库伦修正后的模型进行能量优化,即计算本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同ag掺杂浓度cd-ag共掺杂
zno单层的光学特性:
221.本实例计算的光学特性是指计算介电函数的虚部和光吸收系数,其计算如下:
222.7.6.1)在线性响应范围内,半导体的宏观光学响应函数可用复介电函数ε(ω)表示为:
223.ε(ω)=ε1(ω)+jε2(ω),
224.其中ω是入射光子频率,ε1(ω)和ε2(ω)分别为介电函数实部和虚部,j表示虚部单位;
225.7.6.2)根据占据态和非占据函数的矩阵元素及kramers-kronig色散关系,得到介电函数虚部ε1(ω)和实部ε2(ω):
[0226][0227][0228]
其中e是电子电量,m为自由电子质量,c和v分别为导带和价带,k为倒格矢,bz表示第一布里渊区,δ为狄拉克函数,e
c
(k)和e
v
(k)分别为导带和价带的本征能级,|e
·
m
cv
(k)|表示动量跃迁矩阵元,是约化普朗克常量;
[0229]
7.6.3)根据ε1(ω)和ε2(ω),计算得到吸收系数α(ω):
[0230][0231]
上述计算的本征zno单层、cd单掺杂zno单层,ag单掺杂zno单层和不同ag掺杂浓度cd-ag共掺杂zno单层的介电函数虚部ε2(ω)以及光吸收系数α(ω),如图9所示,其中:
[0232]
图9(a)是本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同ag掺杂浓度cd-ag共掺杂zno单层的介电函数虚部,该图中横坐标表示能量,纵坐标表示介电函数虚部,从图中9(a)可以看出无论是cd-zno,ag-zno还是不同ag掺杂浓度下cd-ag共掺杂zno-ml在大于5.0ev的高能区域,介电函数虚部基本没有发生变化,主要是由于在高能方向上的跃迁主要来自zn-3d和o-2s态之间的电子跃迁产生,这表明掺杂元素主要作用在低能区域。相较于本征zno,不同掺杂浓度的cd-ag共掺杂峰值均向低能方向移动,整体图谱显示为红移。ag-zno、cd-ag-zno,cd-3ag-zno的介电函数峰值位于0.2ev附近,这主要来自于ag-4d和o-2p之间的电子跃迁,而对于cd-2ag-zno在介电函数峰值在0.8ev,这个峰值的变化主要是由cd-2ag-zno是间接带隙半导体,相较于cd-ag-zno体系发生蓝移,而之后随着ag原子的掺入,cd-3ag-zno的介电函数峰又向低能方向移动,发生红移,与本征zno相比,不同ag掺杂浓度下的cd-ag共掺杂zno介电函数表现为红移。
[0233]
图9(b)是本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同ag掺杂浓度cd-ag共掺杂zno单层的光吸收系数,该图中横坐标表示能量,纵坐标表示光吸收系数,从图9(b)可以看出,在可见光范围1.6-3ev内,本征zno单层的光吸收系数为0。ag-zno和cd-ag-zno,cd-2ag-zno,cd-3ag-zno体系的吸收边均位于0.01ev附近,相较于本征zno单层,发生了红移,在可见光范围的响应得到增强。与本征zno相比,cd-ag-zno体系光吸收系数增加,这主要是由于cd和ag原子的协同作用使得禁带宽度减小,此外ag原子的掺入在禁带中引入
了杂质能级,在入射光照射后,价带的电子只需要较少的能量就可以通过杂质能级跃迁到导带,体系的光吸收系数得到提升;并且随着ag掺杂浓度的增大,cd-2ag-zno,cd-3ag-zno体系的光吸收系数也得到了进一步的增加,cd-3ag-zno的提升最为明显,主要原因是是禁带宽度减小以及多条杂质能级的出现。由此可见ag元素能有效提高cd-ag共掺杂zno单层在可见光范围的光吸收系数,增强zno单层的光催化能力。
[0234]
7.7)对步骤5几何优化后的模型的各个原子的电子轨道增加一个能量项进行库伦修正,即zn的3d轨道、o的2p轨道、cd的4d轨道、ag的的4d轨道增加的能量项分别是10ev,7ev、2ev、5ev,得到库伦修正后的模型;
[0235]
7.8)采用广义梯度近似加上库伦修正的方法对库伦修正后的模型进行能量优化,计算得到的不同cd掺杂浓度cd-ag共掺杂zno单层的能带结构如图10所示,其中:
[0236]
图10(a)是cd-ag-zno’的能带结构,该图中给出了自旋向上和自选向下的能带结构,该图中e
f
表示费米能级,从图中可以看出自旋极化向上和向下的禁带宽度分别为3.683ev,3.787ev。与ag-zno相比,掺杂体系的禁带宽度进一步减小。
[0237]
图10(b)是2cd-ag-zno的能带结构,该图中给出了自旋向上和自选向下的能带结构,该图中e
f
表示费米能级,从图中可以看出自旋极化向上和向下的禁带宽度为3.510ev,3.749ev。
[0238]
图10(c)是3cd-ag-zno的能带结构,该图中给出了自旋向上和自选向下的能带结构,该图中e
f
表示费米能级,从图中可以看出自旋极化向上和向下的禁带宽度为3.488ev,3.647ev。综上,随着cd掺杂浓度的增加,cd-ag共掺杂体系的禁带宽度减小,由此可见cd元素能够有效的调整cd-ag共掺杂zno单层体系的禁带宽度。
[0239]
从图10(a)-图10(c)中可以看出,不同cd掺杂浓度cd-ag共掺杂zno单层的自旋能带结构不对称,表明掺杂体系都具备磁性。
[0240]
7.9)对步骤5几何优化后的模型的各个原子的电子轨道增加一个能量项进行库伦修正,即zn的3d轨道、o的2p轨道、cd的4d轨道、ag的的4d轨道增加的能量项分别是10ev,7ev、2ev、5ev,得到库伦修正后的模型;
[0241]
7.10)采用广义梯度近似加上库伦修正的方法对库伦修正后的模型进行能量优化,计算得到的不同cd掺杂浓度cd-ag共掺杂zno单层的态密度图如图11所示,其中:
[0242]
图11(a)-11(c)分别是cd-ag-zno’,2cd-ag-zno,3cd-ag-zno的态密度图,该图中给出了自旋向上和自旋向下的总态密度tdos,自旋向上和自旋向下的分态密度pdos,从图11(a)-11(c)中可以看出导带主要是由zn-4s,cd-5s,ag-5s和o-2p共同贡献,价带主要是zn-3d,o-2p,cd-4d和ag-4d共同贡献。随着cd掺杂浓度增加,导带底下移,价带顶没有发生变化,这主要是由于cd掺杂浓度的增加,cd-4s态增加,导带中的s-s轨道成键的相互作用增强,使得导带向低能方向移动,从而引起禁带宽度减小。随着cd掺杂浓度增大,-6ev附近cd-4d的态密度峰值增加,与o-2p之间的作用增强,使得总态密度峰值增加。
[0243]
7.11)对步骤1、步骤2、步骤4、步骤5各步骤几何优化后的模型的各个原子的电子轨道增加一个能量项进行库伦修正,即zn的3d轨道、o的2p轨道、cd的4d轨道、ag的的4d轨道增加的能量项分别是10ev,7ev、2ev、5ev,得到库伦修正后的模型;
[0244]
7.12)采用广义梯度近似加上库伦修正的方法对库伦修正后的模型进行能量优化,计算本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同cd掺杂浓度cd-ag共掺杂
zno单层的光学特性;
[0245]
本实例计算的光学特性是指计算介电函数的虚部和光吸收系数,其计算如下:
[0246]
7.12a)在线性响应范围内,半导体的宏观光学响应函数可用复介电函数ε(ω)表示为:
[0247]
ε(ω)=ε1(ω)+jε2(ω)
[0248]
其中ω是入射光子频率,ε1(ω)和ε2(ω)分别为介电函数实部和虚部,j表示虚部单位;
[0249]
7.12b)根据占据态和非占据函数的矩阵元素及kramers-kronig色散关系,得到介电函数虚部ε1(ω)和实部ε2(ω):
[0250][0251][0252]
其中e是电子电量,m为自由电子质量,c和v分别为导带和价带,k为倒格矢,bz表示第一布里渊区,δ为狄拉克函数,e
c
(k)和e
v
(k)分别为导带和价带的本征能级,|e
·
m
cv
(k)|表示动量跃迁矩阵元,是约化普朗克常量;
[0253]
7.12c)根据ε1(ω)和ε2(ω),计算得到吸收系数α(ω):
[0254][0255]
上述计算得到的本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同cd掺杂浓度cd-ag共掺杂zno单层的介电函数虚部ε2(ω)以及光吸收系数α(ω)如图12(a)-12(b)所示。
[0256]
图12(a)是本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同cd掺杂浓度cd-ag共掺杂zno单层的介电函数虚部,该图中横坐标表示能量,纵坐标表示介电函数,从图12(a)中可以看出对于不同cd掺杂浓度下的cd-ag共掺杂体系,在0.2ev附近出现一个新的介电函数峰值,这个峰值主要来自ag-4d与o-2p轨道之间的电子跃迁,并且随着cd掺杂浓度的增加,介电函数峰值增加;
[0257]
图12(b)是本征zno单层、cd单掺杂zno单层、ag单掺杂zno单层和不同cd掺杂浓度cd-ag共掺杂zno单层的光吸收系数,该图中横坐标表示能量,纵坐标表示光吸收系数,从图12(b)可以看出ag单掺杂和不同cd掺杂浓度cd-ag共掺杂的吸收边都位于0.01ev范围,与本征zno-m相比,吸收边红移,并且随着cd掺杂浓度的增大,在0-1.5ev红外光范围内,cd-ag共掺杂zno单层吸收系数增大,zno单层的光催化能力得到了进一步的提升,这主要是由于禁带宽度的减小;
[0258]
7.13)通过对步骤6得到的形成能和7.1)-7.12)中得到的能带结构图,态密度图和光学特性的分析对比,得到cd-ag共掺杂可以提高zno单层的光催化特性。
[0259]
综上,cd-ag共掺杂zno单层的光吸收系数得到很大的增强,从而有利于增强zno单层的光催化活性,为zno单层半导体催化性能的研究和发展提供了理论依据,使得zno单层能够很好地应用于光催化领域,实现对于污水进行有效的处理。
技术特征:
1.一种基于cd-ag共掺杂改善zno单层光催化特性的计算方法,其特征在于,包括如下:1)将zno体材料原胞沿x轴和y轴构成的平面进行切片,在切片后的zno材料z轴方向上添加真空层得到zno单层,并对该zno单层进行几何优化,得到本征zno单层模型;2)在本征zno单层模型中掺杂cd原子,得到cd单掺杂zno单层模型,并采用广义梯度近似法对该模型进行几何优化计算体系总能量e
total
,得到优化后的cd单掺杂zno单层;3)在cd单掺杂zno单层模型的不同掺杂位置掺杂不同浓度的ag,并进行几何优化和总能量e
total
选取,得到不同ag掺杂浓度的cd-ag共掺杂zno单层模型;4)在本征zno单层模型中掺杂ag原子,得到ag单掺杂zno单层模型,并采用广义梯度近似法对该模型进行几何优化计算体系总能量e
total
,得到优化后的ag单掺杂zno单层模型;5)在ag单掺杂zno单层模型的不同掺杂位置掺杂不同浓度的cd,并进行几何优化和总能量e
total
选取,得到不同cd掺杂浓度的cd-ag共掺杂zno单层模型;6)根据上述2)、3)、4)、5)各步优化后的总能量e
total
,计算掺杂体系的形成能e
form
;7)采用密度泛函理论结合平面波赝势方法对1)、2)、3)、4)、5)各步几何优化后的模型进行仿真计算,得到能带结构,态密度和光学特性:7a)对1)、2)、3)、4)、5)各步几何优化后的模型的各个原子的电子轨道增加一个能量项进行库伦修正,得到库伦修正后的模型,采用广义梯度近似法加上库伦修正的方法对库伦修正后的的模型进行能量优化,计算得到本征和掺杂zno单层的能带结构图,态密度图和光学特性;7b)通过对6)中计算的形成能e
form
和7a)中得到的能带结构图,态密度图和光学特性的分析对比,得到通过cd-ag共掺杂提高zno单层的光催化特性。2.根据权利要求1所述的方法,其特征在于,1)中对zno单层进行几何优化,其实现如下:1a)设置参数:包括描述和修正交换关联能的近似方法和修正泛函、描述价电子与原子核之间的相互作用的赝势、k点网格、布里渊区k点的路径、平面波能量精度、原子之间相互作用力、晶体的内应力、原子的最大位移、自洽收敛误差、平面波截断能量;1b)对zno单层的晶格常数和原子位置同时进行优化:1b1)选择优化超胞,设置迭代次数为100;1b2)在上述参数条件下进行第一步迭代,计算zno单层的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面;1b3)对势能面求最低极小值,由于此时该值大于设置的收敛精度,则该步迭代结束,改变晶格常数或移动原子位置进行1b4)迭代计算;1b4)重新计算zno单层的总势能,并利用总势能随原子的不同坐标参数变化得到的图像构成势能面,对势能面求最低极小值,将该值与设置的能量的收敛精度进行比较:若该值小于设置的收敛精度,则整个迭代过程自动中断,几何优化结束;若该值大于设置的收敛精度,则该步迭代结束,执行1b5);1b5)改变zno单层的晶格常数或移动原子位置进行下一步迭代计算,重复该步操作,直到系统在设置的迭代次数内,检测到势能面的最低极小值小于设定的能量收敛精度时视为收敛,整个迭代过程自动中断,几何优化结束。3.根据权利要求1所述的方法,其特征在于,2)中采用广义梯度近似法对cd单掺杂zno
模型进行几何优化,其实现过程与zno单层模型的优化过程相同,只是该优化过程中优化的模型是cd单掺杂zno单层模型。4.根据权利要求1所述的方法,其特征在于,所述3)的具体实现如下:3a)在cd单掺杂zno单层模型上,选择在距cd原子为近邻、次近邻、远近邻三种不同掺杂位置上掺入ag原子,得到三种不同的cd:ag=1:1的cd-ag共掺杂zno单层模型,并对其采用广义梯度近似法进行几何优化计算模型的总能量,选取总能量最低的模型;3b)在选取总能量最低的cd:ag=1:1的cd-ag共掺杂zno单层模型上,选择在距该模型确定的ag原子为近邻、次近邻、远近邻三种不同掺杂位置上掺入ag原子,得到三种不同的cd:ag=1:2的cd-ag共掺杂zno单层模型,并对其采用广义梯度近似法进行几何优化计算模型的总能量,选取总能量最低的模型;3c)在选取总能量最低的cd:ag=1:2的cd-ag共掺杂zno单层模型上,选择在距该模型确定的ag原子为近邻、次近邻、远近邻三种不同掺杂位置上掺入ag原子,得到三种不同的cd:ag=1:3的cd-ag共掺杂zno模型,并对其采用广义梯度近似法进行几何优化计算模型的总能量,选取总能量最低的模型,得到不同ag掺杂浓度的cd-ag共掺杂zno单层模型。5.根据权利要求1所述的方法,其特征在于,3)中对于不同ag掺杂浓度的cd-ag共掺杂zno单层模型进行几何优化,其实现过程与zno单层模型的几何优化过程相同,只是该优化过程中优化的模型是不同掺杂ag浓度下的cd-ag共掺杂zno单层模型。6.根据权利要求1所述的方法,其特征在于,4)中采用广义梯度近似法对ag单掺杂zno单层模型进行几何优化,其实现过程与zno单层模型的几何优化过程相同,只是该优化过程中优化的模型是ag单掺杂zno单层模型。7.根据权利要求1所述的方法,其特征在于,所述5)的具体实现如下:5a)在ag单掺杂zno单层模型上,选择在距ag原子为近邻、次近邻、远近邻三种不同掺杂位置上掺入cd原子,得到三种不同的cd:ag=1:1的cd-ag共掺杂zno单层模型,并对其采用广义梯度近似法进行几何优化计算模型的总能量,选取总能量最低的模型;5b)在选取总能量最低的cd:ag=1:1的cd-ag共掺杂zno单层模型上,选择在距该模型确定的cd原子为近邻、次近邻、远近邻三种不同掺杂位置上掺入cd原子,得到三种不同的cd:ag=2:1的cd-ag共掺杂zno单层模型,并对其采用广义梯度近似法进行几何优化计算模型的总能量,选取总能量最低的模型;5c)在选取总能量最低的cd:ag=2:1的cd-ag共掺杂zno单层模型上,选择在距该模型确定的cd原子为近邻、次近邻、远近邻三种不同掺杂位置上掺入cd原子,得到三种不同的cd:ag=3:1的cd-ag共掺杂zno单层模型,并对其采用广义梯度近似法进行几何优化计算模型的总能量,选取总能量最低的模型,得到不同cd掺杂浓度cd-ag共掺杂zno单层模型。8.根据权利要求1所述的方法,其特征在于,5)中不同cd掺杂浓度的cd-ag共掺杂zno单层模型的几何优化,其实现过程与zno单层模型的几何优化过程相同,只是该优化过程中优化的模型是不同掺杂cd浓度下的cd-ag共掺杂zno单层模型。9.根据权利要求1所述的方法,其特征在于步骤6)中根据上述2)、3)、4)、5)各步优化后的总能量e
total
计算掺杂体系的形成能e
form
,通过如下公式计算:e
form
=e
total-e
zno
+∑n
i
μ
i
,其中,e
total
是掺杂体系总能量,e
zno
表示的是本征zno单层的总能量,n
i
表示的是掺杂体
系中得到或失去的原子个数,当n
i
小于零表示掺杂体系掺入原子,n
i
大于零表示掺杂体系失去原子,i表示的是掺入或失去的原子,μ
i
表示的是对应原子的化学势。10.根据权利要求1所述的方法,其特征在于步骤7a)中得到本征和掺杂zno单层的光学特性,包括介电函数虚部和光吸收系数,其计算如下:7a1)在线性响应范围内,半导体的宏观光学响应函数可用复介电函数ε(ω)表示为:ε(ω)=ε1(ω)+jε2(ω),其中ω是入射光子频率,ε1(ω)和ε2(ω)分别为介电函数实部和虚部,j表示虚部单位;7a2)根据占据态和非占据函数的矩阵元素及kramers-kronig色散关系,得到介电函数实部ε1(ω)和虚部ε2(ω):(ω):其中e是电子电量,m为自由电子质量,c和v分别为导带和价带,k为倒格矢,bz表示第一布里渊区,δ为狄拉克函数,e
c
(k)和e
v
(k)分别为导带和价带的本征能级,|e
·
m
cv
(k)|表示动量跃迁矩阵元,是约化普朗克常量;7a3)根据ε1(ω)和ε2(ω),计算得到吸收系数α(ω):
技术总结
本发明公开了一种基于Cd
技术开发人、权利持有人:杨涵 吴茜 杨婷 刘妍 王平

